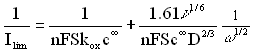 (1)
(1)Films of poly(N-vinylcarbazole) (PVCz) were prepared and studied in the presence of different cations using spectroelectrochemical and electrochemical quartz crystal microbalance methods. Evidence for effect of the cation on both formation and redox behaviour has been obtained. The experimental results have been interpreted by considering the presence of different types of ionic species inside the polymers layer (free anion, screened anion, bound anion and cation).
Keywords:Poly(N-vinylcarbazole), Electropolymerisation, Spectroelectrochemistry, EQCM, Cation effect.
In recent years, the formation and characterisation of conducting and semi-conducting polymers have received increasing attention mainly due to the possibility of controlling and modifying their electrical conductivity. The interest of studying electrically conducting polymers lies primarily in their wide field of applications : e.g. fuel cell [1], polymer modified electrodes [2], sensors and biosensors [3], electronic and electrochromic devices [4], conducting matrices and membranes [5], protection layers [6], and electrocatalytic materials [7]. As a result, most investigations have been undertaken for preparative purposes. More recently, studies became focused on the fundamental aspects of the initial stages of electrodeposition [8-13].
Poly(N-vinylcarbazole) (PVCz), which displays interesting electrical properties has already been studied extensively [14-19] because of possible application as a light emitting diode material [20], a battery material [21] and in electrochromic devices [22]. It is generally accepted that the electropolymerisation of N-vinylcarbazole proceeds through an ECE mechanism [23]. The monomer oxidation leads to the formation of a radical cation which reacts with another oxidised monomer. Then, the dimer is oxidised and couples with another oxidised monomer. The growing polymer is simultaneously oxidised because of its oxidation potential which is less positive than that of the monomer. As a result, radical cations, responsible for conductivity of the film, are created along the polymer chain. The charge may be transferred through the film. Therefore, oxidation of additional monomer units at the polymer˝solution interface still occurs leading to the growth of the polymer film. The electroneutrality of the system is achieved by the incorporation into the polymer matrix of anions of the supporting electrolyte. However, it has been reported in a few recent papers, dealing with other polymers [24-26], that the overall process is more complex and that also the cations of the supporting electrolyte play an important role, and several models have been proposed to describe this peculiar behaviour [27,28].
In this work, we present the results of the studies on the role of the cations on the formation and the electrochemical behaviour of poly(N-vinylcarbazole) films by means of electrochemical, spectroelectrochemical and quartz crystal microbalance methods.
2. ExperimentalThe electrochemical experiments were performed in a single compartment cell with a conventional three electrode configuration with a platinum gauze as the counter electrode and a double junction reference electrode Ag 0.1 mol dm-3 AgNO3 in acetonitrile. All potentials are referred to this electrode. The electrochemical measurements were carried out by means of AUTOLAB (Eco Chemie, Netherlands).
Poly(N-vinylcarbazole) films were deposited under potentiostatic conditions on a stationary or rotating platinum disc electrodes (RDE Tacussel with an area 0.038 cm2), indium tin oxide coated glass (ITO with an area 2.8 cm2) or gold electrodes (for EQCM) from acetonitrile solutions containing 0.01 mol dm-3 N-vinylcarbazole (Fluka) and 0.5 mol dm-3 LiClO4 (Aldrich) or N(C4H9) 4ClO4 (TBAClO4, Fluka) as supporting electrolytes. Previous studies on polymerisation of PVCz carried out on various substrates [12,16,29] showed that the type of the substrate does not influence the electroactivity and morphology of the films provided that the other polymerisation conditions are kept the same.
Acetonitrile (Fluka) was dried by adding an activated alumina powder for a few days before the measurements. The platinum disc was polished with a diamond paste (0.25 µm) and rinsed with pure acetonitrile before polymerisation. The ITO electrodes were washed with acetone in an ultrasonic bath for several minutes.
Optical spectra were recorded using a double beam UV-visible spectro-photometer (Lambda 12, Perkin-Elmer). The cell for in-situ spectroelectrochemical measurements contained the ITO working electrode and miniaturised reference and counter electrodes. The blank cell contained an uncoated ITO electrode in acetonitrile solution of the supporting electrolyte.
The EQCM experiments were performed by means of a nanobalance type M103A (UELKO, Poland), combined with AUTOLAB (Eco Chemie, The Netherlands). The working electrodes for EQCM measurements were 10 MHz AT-cut quartz crystals with evaporated gold electrodes (International Crystal Manufacturing Co, Ltd., Oklahoma City, OK) with piezoelectrically and electrochemically active areas of about 0.21 cm2 and 0.23 cm2, respectively.
3. Results 3.1. Influence of the cations on the electropolymerisation of N-vinylcarbazole.The influence of cations on the regime of polymerisation of PVCz was studied on the large area (2.8 cm2) ITO electrodes. The current transients presented in Fig.1a suggest that the rate of the process is much lower in the solution containing TBAClO4 than in that with LiClO4. According to the nucleation and growth models [30,31], the mathematical analysis of the time dependence of an early part the current transient allows the regime of the process to be established. Using the procedure proposed by Hillman and Mallen [32], we plotted the experimental data in the form of a ln (I-I0) vs. ln (t-t0) graph (Fig.1b) within the time range where the overlap of the growing centres or diffusion zones may be neglected, i.e. for increasing part of the current transient. We obtained a slope of 2 for the early linear part of the graph which may be characteristic of two possible mechanisms, progressive nucleation and two-dimensional growth (PN2DG) or instantaneous nucleation and three-dimensional growth (IN3DG) [31]. As the electrodeposition proceeds, the slope of the plots presented in Fig.1b changes from 2 to 1/2. The latter is characteristic of the growth controlled by the hemispherical diffusion to the growing centres. One may note, that the duration of each part of the transient is markedly longer in the solution of VCz+TBAClO4 than in VCz+LiClO4.
3.1.1 Determination of the kinetic parameters of electrodeposition by means of RDE experimentsThe RDE experiments were carried out at various rotation rates (w = 2pf) and at different potentials of electropolymerisation in both deposition solutions (VCz+LiClO4 and VCz+TBAClO4). As it is well known from the literature [10,22,33], the polymerisation of VCz may occur via the benzene rings as well as through the vinyl groups. The former coupling leads to the formation of a green poly(3,3'N-vinylcarbazole) conducting film on the working electrode, whereas the latter results in precipitation of non-conducting poly(9,9'N-vinylcarbazole) in the form of a white deposit which does not stick to the electrode. Both processes occur simultaneously during the first few minutes of electrodeposition especially in LiClO4-containing solution. Therefore, in order to avoid the disturbing effect of poly(9,9'N-vinylcarbazole), we used the prepolymerisation procedure described elsewhere [29]. The charge consumed during prepolymerisation was about 1.5 C.
The previous studies on electrodeposition of PVCz on RDE in VCz+LiClO4 solution have shown that the shape of the current transients depends both on the height of the potential step and the rotation rate [12]. It has also been demonstrated that the increase of the potential step amplitude results in the change of the rate limiting step of the process from a kinetic to a diffusion control. As is shown in Fig.2, the change of the supporting electrolyte from LiClO4 to TBAClO4 also results in the change of the rate limiting step, especially at the low rotation rates. A linear extrapolation of the resulting Ilim vs. w1/2 curves from w1/2= 4 rad1/2 s-1/2 to zero is only possible in the case of the electrodeposition taking place in LiClO4. A deviation of the results obtained in TBAClO4 from the straight lines that intersect the origin of the graph supports the fact that in this case the process is much slower and the reaction is mainly kinetically controlled.
Therefore, we plotted the experimental data in the form of a Levich-Koutecky plot, (Ilim)-1 vs. w-1/2 (Fig. 3). According to the Levich-Koutecky expression :
(1)For the process carried out in NBu4ClO4, the Levich-Koutecky plot gives a linear relationship. This allows us to obtain a value of the diffusion coefficient of the monomer (2.55´10-5 cm2 s-1). From the dependence of log kox vs. E presented in Fig.4, we also determined a value of bn (0.43), where b is a transfer coefficient for anodic reaction. In calculations we assumed that one electron is involved in oxidation of one monomer unit. The value of bn suggests that this assumption is reliable. In order to justify the value of n we also studied the oxidation of the monomer on a stationary electrode by means of cyclic voltammetry at different scan rates. The shape of the I-E curves and the shift of the anodic peak with increasing scan rate u, suggest the irreversible charge transfer process [35]. We calculated bn from these data and the obtained value is in agreement with the result derived from RDE experiments. We put the obtained values of D, bn and n=1 to the equation for the peak current: Ip = (2.99´105)n(bn)1/2ScD1/2u1/2. The calculated value of Ip differs only by about 3% from the experimental one supporting, that our assumption concerning the value of n was correct.
In order to gain more data on the influence of the cations on the electrodeposition of PVCz, we also made a series of experiments by means of EQCM and spectroelectrochemical methods. The corresponding results are presented and discussed below.
3.1.2 EQCM studies of electropolymerisationIn both deposition solutions the polymerisation was carried out at the potential of 0.7 V. The previous studies performed in VCz+LiClO4 by means of crystal admittance measurements showed that the PVCzLiP films obtained under such conditions are rigid and that the polymerisation proceeds with 100% efficiency [29]. In such case, the Sauerbry equation [36], which for the 10 MHz AT-cut quartz crystals may be written in the form:
may be used to convert the changes of the frequency into the mass changes at the interface.
The changes of Df as a function of the charge density, Qdep, passed through the interface during deposition of PVCz in both electrolytes are presented in Fig.5a. It is surprising that a linear relationship (Df vs. Qdep) was obtained only with LiClO4 as supporting electrolyte (curve 1). From the slope of this graph, we may calculate the molar mass equivalent of deposited species (including dopant ions), according to the equation:
where n is the number of electrons involved. In these calculations, we used n = 2.45, where 2 electrons are associated with polymerisation and 0.45 electrons are associated with doping of one monomer unit in PVCz. The obtained value, 245 g mol-1, is a little higher than the sum (equal to 242.7 g mol-1) of the molar mass of monomer and 0.5 of dopant ion (ClO4-) per monomer unit being incorporated into the film.
In the case of electrodeposition carried out in TBAClO4, the plot of df vs. Qdep is non-linear. This fact may be indicative of the non-rigidity of the film or of the low efficiency of the growth process. The latter case means that the charge passed through the interface may be consumed also in another reaction than oxidation of the monomer or that the process of attachment of oxidised monomer to the growing polymer chains is very slow and part of oxidised monomer units remains in the solution.
3.1.3. Spectroscopic studies of polymerisation of VCzThe absorbance spectrum of PVCz in the oxidised state shows a peak at about 400 nm followed by a broad band around 900 nm. In the literature [23], the peak at 400 nm is ascribed to the absorption of the radical cation and the second band is attributed to dications and/or dicarbazyl perchlorate salt. It has been shown however [16], that the spectra of poly(N-vinylcarbazole) do not exhibit any isosbestic point typical of the systems with oxidation of polarons to bipolarons (dications) i.e. in this case only the polarons are responsible for conductivity. The band at 400 nm was selected in order to monitor the changes of absorbance during the potentiostatic deposition of PVCz. The electropolymerisation conditions were kept the same as in the EQCM experiments. The results are plotted in Fig.5b in the form of absorbance (A) vs. Qdep plots. It is interesting to note that the same general trend is obtained in both Figs 5a and 5b: a linear relation for LiClO4 and non-linear, with the presence of two distinct segments, for TBAClO4. The change of the slope observed for TBAClO4 occurs nearly at the same charge density as that when the curvature starts in Df vs. Qdep plot, confirming that this charge may correspond to the change in the morphology of the polymer or/and to the change in the electrodeposition efficiency. As it has been mentioned above, if the rate of the film growth on the substrate is low, part of the oxidised monomers may remain in the solution. By recording the absorbance spectra of the solutions after polymerisation, we have observed a shoulder at about 400 nm in the spectrum of the polymerisation bath containing TBAClO4, while in the case of the solution containing LiClO4 no significant increase of absorbance in this wavelength range was detected. These experimental data indicate the presence of radical cations in the TBAClO4 solution after polymerisation and support that in this case the efficiency of the film formation is indeed lower than 100%.
From the results presented in Fig.5a and Fig.5b, one can correlate the absorbance with the mass changes for the same polymerisation charge densities. It follows from this correlation that the same absorbance of the films corresponds to a markedly higher change of the resonant frequency when the electrodeposition is carried out from TBAClO4 solution than from LiClO4 one. For example, the films of absorbance 0.3 are obtained after passage of about 0.047 C cm-2 in TBAClO4 and only about 0.015 C cm-2 in LiClO4. These values of charge densities correspond to changes of the resonant frequency -5.2 kHz in TBAClO4 and -3.5 kHz in LiClO4. Therefore, it seems that in the former case with the lower polymerisation efficiency, the polymer film accommodates the species of higher molar mass.
According to Hillman et al. [37], when the polymerisation occurs with 100% efficiency, the absorbance (A) may be related to the polymerisation charge density (Qdep) by means of equation :
where e is the molar absorption coefficient and n is the number of electrons involved in the polymerisation/oxidation of each monomer unit. The value of e determined by means of this equation for PVCz film obtained in the presence of LiClO4 is equal to 4790 mol-1 l cm-1.
3.2. Influence of the cations on the redox behaviour of PVCz filmsThe charge passed during the electropolymerisation is often used to make an estimation of the film thickness, provided the polymerisation occurs with 100% efficiency. This is the case only for polymerisation carried out in LiClO4. Therefore, in order to compare quantitatively the experimental data obtained in different supporting electrolytes, we decided to stop the electropolymerisation on ITO when the absorbance measured at 400 nm reaches a given value kept equal in both supporting electrolytes. However, the quantitative comparison of the films obtained in both electrolytes for EQCM studies is not so easy, because as shown above, the same change of the resonant frequency does not denote the same electroactivity of the films.
After deposition, the films were firstly investigated in the same supporting electrolyte as the one used for electropolymerisation before being transferred to the solution containing the electrolyte with a different cation, i.e. from LiClO4 to TBAClO4 or from TBAClO4 to LiClO4 (at the same concentration, 0.5 mol dm-3) and studied again using the same procedure. Before each change of the solution, the films were rinsed with dry acetonitrile.
3.2.1. Comparison of the redox behaviour of PVCz films obtained in the presence of different electrolyte cationsTwo fresh polymer layers were obtained potentiostatically in different supporting electrolytes at a polymerisation potential of 0.7 V. The films were firstly crosslinked by cycling the potential in the range 0 V Ž 0.75 V Ž 0 V in the corresponding monomer free solutions until the voltammograms and df vs. E curves reached a reproducible patterns. As may be seen in Fig.6a and Fig.6b, the shape of the cyclic voltammograms of the fresh films obtained in different supporting electrolytes is similar in spite of different current densities. After the crosslinking, the polymers were oxidised and reduced several times by applying a double potential step (from -0.2 V to +0.65 V and back to -0.2 V) and finally, the electroactivity of the films was checked again by means of cyclic voltammetry at various scan rates.
It may be seen from Fig.6a that the electrochemical behaviour of the film obtained and studied in LiClO4 (film PVCzLiP, dashed curve) remained nearly unchanged with respect to that of the fresh film. In contrast, the shape of the voltammogram for the film obtained and studied in TBAClO4 (film PVCzTBAP) is strongly changed and the current density is lowered (dashed curve in Fig.6b). The similar shape of the cyclic voltammograms was also observed when the film PVCzLiP was transferred into TBAClO4 solution (Fig.7a).
In order to compare the change of the film electroactivity quantitatively as a result of the redox treatment in different electrolytes, we calculated the surface concentrations of electroactive centres (G, in mol cm-2) in the films before and after changing the electrolyte, using the equation: G = Qred/nF, where Qred is the charge density consumed during reduction of the film. Qred was determined by integration of the cathodic branch of voltammograms recorded in the potential range from 0.65 V to -0.2V. We used the negative scan in order to avoid the influence of the "overoxidation" process, responsible for the increase of the anodic current observed after the oxidation peak (see Fig.7). The obtained results are listed in Table 1. In this Table, we also compare the surface concentrations of the electroactive centres that are reduced in "crosslinked" films obtained in the two different supporting electrolytes but in these experiments the potential range was broader (from 0.75 V to -0.2 V).
In the case of the redox reaction of PVCzLiP, the curves of Df vs. Q may be used to determine the molar equivalent of the species exchanged at the polymerçsolution interface, by means of the equations (2) and (3). For calculations we used the data corresponding to the cathodic branch of curve at the scan rate 40 mV s1, that allows to avoid the influence of the slow exchange of the solvent or salt [29] as well as overoxidation process. The obtained value, 101(ą1) g mol-1, is close to that corresponding to ClO4-, i.e. 99.5 g mol-1.
The cyclic voltammograms (Fig.7a) and the Df vs. E curves (Fig.8a) recorded for the PVCzLiP film after transferring into a TBAClO4 solution become similar to those obtained for the film prepared and studied in TBAClO4 (curve 1 of Figs 7b and 8b), with anodic wave split into two peaks, at about 0.24V and 0.48 V.
However, the most interesting behaviour was observed when the film deposited and studied in TBAClO4 was transferred into the solution of LiClO4. The first and the second EQCM scans carried out at a low scan rate were not reproducible, with a strong increase of the resonant frequency. The peculiar behaviour was also found under oxidation at low scan rates during the consecutive redox cycles (curves 2 and 3 in Fig.9). The Df vs. E profiles consist of two distinct parts - the first segment with an increase of the resonant frequency (that corresponds to the mass decrease) in the potential range from 0.1 V to 0.48 V and the second one with decrease of Df (i.e. mass increase) for E > 0.48 V. This behaviour is quite different from that recorded for the same film in TBAClO4 (dashed curve in Fig.8b). In the latter case, the polymer mass remained nearly constant up to 0.2 V and then increased. In the corresponding cyclic voltammograms, we observe that the less positive peak (denoted as I in Fig.7b), recorded in TBAClO4 at 0.24 V, shifts towards lower potentials after the change of solution, whereas the second peak (II), at 0.48 V, becomes much sharper. Moreover, there is a small shift of peak II towards less positive potentials during the second scan in LiClO4, whereas peak I vanishes. The described changes in the cyclic voltammograms correspond to an increase of the concentration of the electroactive centres undergoing reduction from 0.5´10-8 mol cm-2 in TBAClO4 to 0.6´10-8 mol cm-2 in LiClO4. It is also noteworthy that in this case the shape of the Df vs. E profiles strongly depends on the scan rate. As can be seen on Fig.9, the increase of the scan rate from 5 mV s-1 to 20 mV s-1 results in the "flattening" of Df vs. E profiles and the curves become looped.
3.2.1b. Spectroelectrochemical measurementsThe poly(N-vinylcarbazole) films were deposited potentiostatically at 0.7 V on the transparent ITO coated glass electrodes. In order to obtain the polymer films containing the same amounts of the oxidised redox centres, we controlled the value of absorbance at 400 nm during polymerisation. A selected absorbance value of 0.7 was reached after passage of 0.032 C cm-2 in the solution of VCz+LiClO4 and 0.082 C cm-2 in VCz+TBAClO4.
Then, the films were rinsed with pure acetonitrile and studied in the corresponding supporting electrolytes. As shown in Fig.10, the general shape of the absorbance spectra of the fresh oxidised as well as reduced films obtained in different supporting electrolytes is similar. However, for wavelengths shorter than 800 nm, the absorbance of the reduced film obtained in TBAClO4 is slightly higher than that of the film obtained in LiClO4. This fact has an effect on the absorbance values of the oxidised PVCzTBAP film and in consequence, a shoulder instead of the peak is obtained at about 400 nm. The increase of absorbance of the reduced PVCzTBAP film became more pronounced when this polymer was oxidised and reduced several times in TBAClO4 (Fig.11). The intensity of the band at 400 nm for the reduced film increased by about 0.043 from step to step whereas it remained nearly constant for the oxidised film.
A comparison of CV and A vs. E behaviour of the films prepared in two different electrolytes is presented in Fig.12. The absorbance values of the films in the oxidised forms are similar but differ when the films are reduced. These results indicate that in spite of nearly the same number of electroactive centres in both cases, the film obtained and studied in TBAClO4 may be reduced only partially.
The results obtained by steps and potential scans in the solutions containing the supporting electrolytes different from those used for polymerisation may be summarised as follows :
Finally, after the redox studies the films were peeled off from the surface and analysed by FTIR. No structural differences between the polymeric film obtained or studied in different supporting electrolytes were found. Therefore, we claim that the polymers did not sustain any irreversible faradaic reaction that influences the electroactivity of the films.
4. DiscussionThe studies of electrodeposition of PVCz films from two electrolytic baths containing different types of cations suggest that the cations affects not only the electroactivity of the films, as it has been reported in the literature [24-28] but also the rate of the polymer growth. This effect is detected from the beginning of the polymerisation. The current transients recorded on the stationary electrodes (Fig.1) indicate that the rate of the film growth is higher in the solution of VCz+LiClO4 than that in VCz+TBAClO4. On the other hand, our spectroelectrochemical results show that the charge passed through the interface to produce the films containing the same amounts of the redox centres has to be markedly higher when the polymerisation is carried out in the presence of TBAClO4 (Fig.5). However, only about 80% of the polarons in the film formed in VCz+TBAClO4 are reduced in the first reduction step even after long polarisation at -0.2 V with respect to the polarons reduced in the film obtained in VCz+LiClO4 (Fig.10). In order to explain these results, we reconsider the model of the polymer matrix. In general, it is accepted that ionic pairs polaron-anion are formed during polymerisation. Such ionic pairs may be immobilised in the polymer matrix and then, the compensation of these species under reduction is achieved by incorporation of cations from the solution. Big cations are not easily accommodated by the film and consequently, the polymer cannot be fully reduced. The change of the size of the electrolyte cation to a smaller one should cause the remarkable increase of the number of the electroactive centres that may be reduced and then again oxidised. However, the data presented in Table 1, in Fig.7 as well as the spectroelectrochemical results do not confirm this hypothesis. The change of the supporting electrolyte from TBAClO4 to LiClO4 results in a small increase of the concentration of reducible centres. But, the absorbance measured at 400 nm for the reduced PVCzTBAP film remains still remarkably higher than that of the film obtained in LiClO4 and studied under the same conditions in both supporting electrolytes. The shape of the cyclic voltammogram changes but is still different from that for the film obtained and studied in LiClO4 (Fig.7). Therefore, it is obvious that we should consider a model of the polymer matrix suggesting that not only anions but also cations are incorporated into the growing films. This may be also consistent with the results presented in Fig.1b. According to Scharifker and Hills [38] during the stage of the film growth controlled by diffusion, the diffusion zones begin to overlap and the replacement of the material at the electrode becomes hindered. As both ionic species (anions and cations) are likely present near the electrode, they may both be trapped inside the polymer matrix. The incorporation of cations into a growing polymer film has already been reported in the literature; the amount of cations (Na+) remaining in poly(3-methylthiophene) film after consecutive polymerisation scans has been detected by radiometric measurements [39]. One may expect that less mobile ions are more slowly replaced by the monomer molecules and slow down the rate of the polymer growth. The lithium cations, in spite of a high degree of solvation, are still more mobile than TBA+ (Stokes radii: Li+ = 3.46 Å, TBA+ = 3.91 Å, and ClO4- = 2.32 Å) [40]. However, the nanogravimetric results obtained for electrodeposition of PVCz from LiClO4 suggest that the ions are incorporated into the polymer matrix without their solvation shells. On the other hand, the EQCM results obtained for polymerisation from TBAClO4 may seem to be a little confusing because the molar mass of TBA+ is much higher than that of Li+ (242 g mol-1 and 7 g mol-1, respectively) whereas the slope of the graph dm vs. dQ is considerably lower than the slope obtained in LiClO4. It may be understood when comparing the corresponding spectroelectrochemical results. As is visible in Fig.5b, in order to obtain the films of the same amount of electroactive centres (polarons), which may be controlled by the value of absorbance at 400 nm, the charge passed during polymerisation has to be markedly higher in TBAClO4 than in LiClO4, probably due to the lower electrodeposition efficiency. At the same time, the corresponding change of the resonant frequency for the film produced in TBAClO4 is much higher than that for the film of the same electroactivity but prepared in LiClO4 (compare Fig.5a and Fig.5b, explained on page 9). We suppose that this difference results from the distinct value of the molar masses of cations, that are also incorporated into the polymer matrix.
In this work, we propose that the reduced polymer matrix is composed of the following species: neutral ("reduced") electroactive centres, oxidised centres trapped inside the film with associated anions and ions from the supporting electrolyte accommodated in a form of ionic pairs or separately. Solvent molecules may be also present in the polymer matrix and the solvent transfer often influences the course of Df vs. E profiles.
The results presented above show that the size of the cation also influences on the redox behaviour of the polymer. As shown in Fig.8, the change of the resonant frequency during the redox scans from 0 V to 0.65 V is about twice as high in the case of the film obtained and studied in LiClO4 as that in TBAClO4. Also about twice as high is the surface concentration of the electroactive centres that are reduced and oxidised in LiClO4 (Table 1). When the polymer obtained in LiClO4 was studied in the solution of TBAClO4, the shape of cyclic voltammograms and Df vs. E profiles became similar to these of the film obtained and studied in TBAClO4 (Fig.7 and Fig.8). The two separated anodic and cathodic peaks present in the cyclic voltammograms may be ascribed to consecutive exchange of ions, i.e. cations at the less positive and anions at more positive potentials. A similar behaviour has been reported in the literature [41,42]. The exchange of both ionic species in the redox behaviour of the polymer has also been postulated for polypyrrole [28,43,44] polythiophenes [44-46] and poly(1-naphthol) [47] films.
In view of the results presented above and the literature data, we also reconsider the overall scheme of the redox process of the PVCz film. One may consider three kinds of anions involved in the whole redox process of the polymer [41] :
We also postulated above that the cations of the electrolyte may be trapped inside the film. Since the overall neutrality of the polymer matrix in the reduced state has to be fulfilled, we consider in the scheme below, that the charge of these is compensated by the charge of anions, and denote it as ![]()
Thus, considering the presence in PVCz film of all the species mentioned above (except the solvent molecules), the redox process of the polymer may be written as follows:
It may be expected that the amount of screened anions, associated with the oxidised centres trapped in a fresh film, should depend on the size of electrolyte cation used for the polymerisation. The big cations incorporated into the polymer matrix may influence on the spatial configuration of the polymer matrix and hinder the outcome of anions under reduction by blocking the paths of ions transport through the film (the ionic ionic radii without solvation: Li+ = 0.6 Å, TBA+ = 3.91 Å). On the other hand, the free anions cannot be neutralised by the cations from the solution if the latter ones are too big to penetrate the film deeply. Changing the cation during the study of the electrochemical behaviour of the different films should mainly affect the population of three species: bound anions, ![]() , screened anions,
, screened anions, ![]() and the "charge compensator" anions
and the "charge compensator" anions ![]() .
.
The spectroelectrochemical results obtained for both types of films support these hypotheses. The value of absorbance of the PVCzLiP film in the oxidised state (i.e. the total number of polarons) was not affected by the type of cation of the supporting electrolyte. In contrast, the absorbance in the reduced state was higher in the electrolyte with a bigger cation (Figs 11-13). This means, that a larger amount of polarons remained trapped in the polymer matrix and in consequence, non-neutralised bound anions become screened anions. In contrast, the transfer of the PVCzTBAP film into the solution with a small cation results in some decrease of the number of screened anions due to the easier penetration of small and mobile Li+ ions in the film.
The question that remains concerns the EQCM profiles recorded just after the transfer of PVCzTBAP film into LiClO4 and during the consecutive redox scans at the low scan rates (Fig. 7b, 8b and Fig. 9). This behaviour may be explained when one considers again the overall scheme of the redox reaction of the polymer. When the PVCzTBAP film is studied in TBAClO4, the expulsion of the cations from the bulk of the film is relatively slow and probably only the superficial cations take part in the redox reaction of the film. When this polymer is submitted to a potential cycling in LiClO4, TBA+ cations have to be replaced by Li+. Since the space occupied by TBA+ is relatively large, the Li+ ions may enter the film without desolvation. Another process that could be also taken into account is the exchange of the cations, according to the reaction :
These both processes are likely responsible for non-reproducible Df vs. E behaviours obtained immediately after the change of the supporting electrolytes.
The course of reproducible Df vs. E profiles depends strongly on the scan rate (Fig.9). On curve 1, for E < 0.48 V, the mass decreases as a result of the difference between expulsion of cations and accommodation of anions during oxidation. At the higher potentials, the income of the anions prevails and therefore the polymer mass increases. This curve exhibits an interesting feature: the mass increase during the forward scan (from 0.48 to 0.65 V) is less important than the mass decrease observed on the back scan (from 0.65 to 0.48 V). If only anions were involved, the mass changes would be of the same amplitude (as shown on curve 3). The difference may be attributed to the expulsion of the solvent which is superimposed to the anion incorporation during the forward scan. If the solvent expulsion is slow and still occurs on the back scan, it may also emphasise the mass decrease during the anion outcome. The slow exchange of the solvent in our opinion is also the reason of gradual change of the shape of Df vs. E curves with increasing scan rate (Fig.9). Unfortunately, because of non-linearity of Df vs. Q behaviour for deposition of the film PVCzTBAP, the nanogravimetric results obtained for this film can be interpreted only qualitatively.
5. ConclusionsThe studies of electrodeposition of PVCz by means of RDE, EQCM and spectroelectrochemical methods demonstrate that the rate of the film growth depends on the electrolyte cation size.
The efficiency of polymerisation is 100% in the solution of VCz+LiClO4 and much lower in VCz+TBAClO4.
Both types of ionic species (anions and cations) are trapped in the polymer matrix during the film formation. The cations inside the film are mainly associated with anions whereas the anions may exist in the form of free anions, bound anions and screened anions. The population of bound anions, screened anions and cations in the film may be modified by the change of the supporting electrolyte used for redox studies of the polymer.
AcknowledgementsThis work was supported by both Polish KBN funds within the project BST/562/16/97 and Belgian (Région wallonne) 96-11063 grant in the frame of a scientific collaboration between the Republic of Poland and Belgium (Région wallonne). V. C. wishes to thank the "Fonds pour la Formation ŕ la Recherche dans l'Industrie et dans l'Agriculture" (F.R.I.A.) for her PhD grant.
References